Quickstart¶
Installation¶
fastcxt requires Python 3.10+ and a CUDA-capable GPU for the Mamba kernels.
uv pip install -e ".[all]"
pip install -e ".[all]"
For CPU-only development (e.g. preprocessing, simulation), install without the GPU dependencies:
uv pip install -e ".[sim,docs,dev]"
End-to-end example¶
Simulate training data with variable sample sizes:
for N in 10 25 50 100 200; do
fastcxt-simulate --scenario constant \
--data-dir ./sims/n${N} --num-ts 200 --n-samples $N
done
This creates 1000 tree sequences total (200 per sample size) in separate subdirectories.
Preprocess into SFS features and TMRCA targets:
fastcxt-preprocess --base-dir ./sims --out-subdir processed \
--extract-trees --max-samples 200
The preprocessor scans sims/ recursively, discovers all .trees files, and
uses each subdirectory name (n10, n25, …) as the scenario label. The
--max-samples 200 flag pads tree topology features to a consistent dimension
so all sample sizes can be batched together.
Train a model:
fastcxt-train --model base --dataset-path ./sims/processed --gpus 0
Run inference from Python:
import fastcxt
from fastcxt.config import PRESETS
from fastcxt.model import FastCxtModel
config = PRESETS["base"]
model = FastCxtModel(config)
# model.load_state_dict(torch.load("checkpoint.pt"))
from fastcxt.translate import translate_from_ts
import tskit
ts = tskit.load("my_data.trees")
means, variances, index_map = translate_from_ts(
ts, model,
pivot_pairs=[(0, 1), (0, 2)],
mutation_rate=1e-8,
device="cuda:0",
)
Build a TimeAtlas for genome-wide results:
from fastcxt.atlas import TimeAtlas
atlas = TimeAtlas()
atlas.add_arm("2L", means, variances, pairs, window_size=2000)
atlas.save("my_atlas/")
# Query later
atlas = TimeAtlas.load("my_atlas/")
m, v = atlas.query_pair("2L", sample_a=0, sample_b=5)
Visualize with geographic context:
# Generate showcase figures with simulated placeholder data
python scripts/plot_atlas_showcase.py --outdir figures/
This generates publication-quality geographic maps, TMRCA landscapes, population heatmaps, selective sweep panels, and a composite dashboard. See Visualization for the full gallery.
Example results¶
The figures below are from a quickstart run: 1000 constant-demography tree
sequences with variable sample sizes (10–200), the base model preset
trained for 20 epochs with Beta-NLL loss on 3 GPUs.
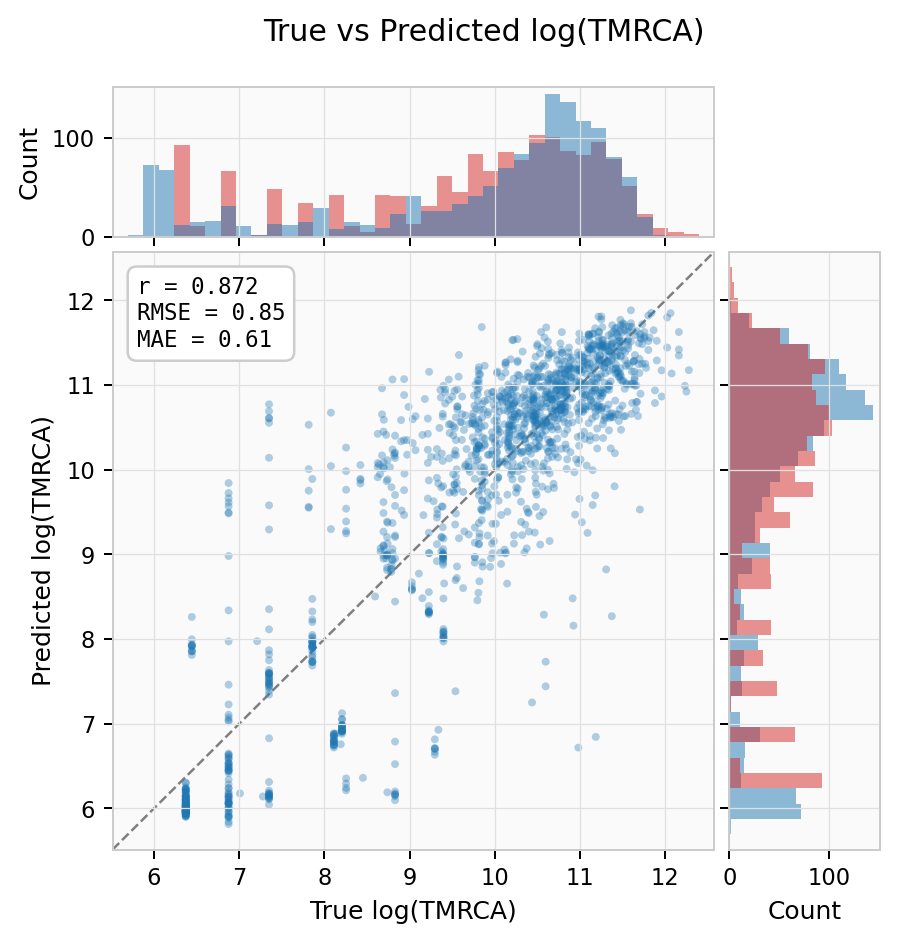
True vs Predicted log(TMRCA) — scatter with marginal histograms showing overall calibration (r = 0.87, RMSE = 0.85):
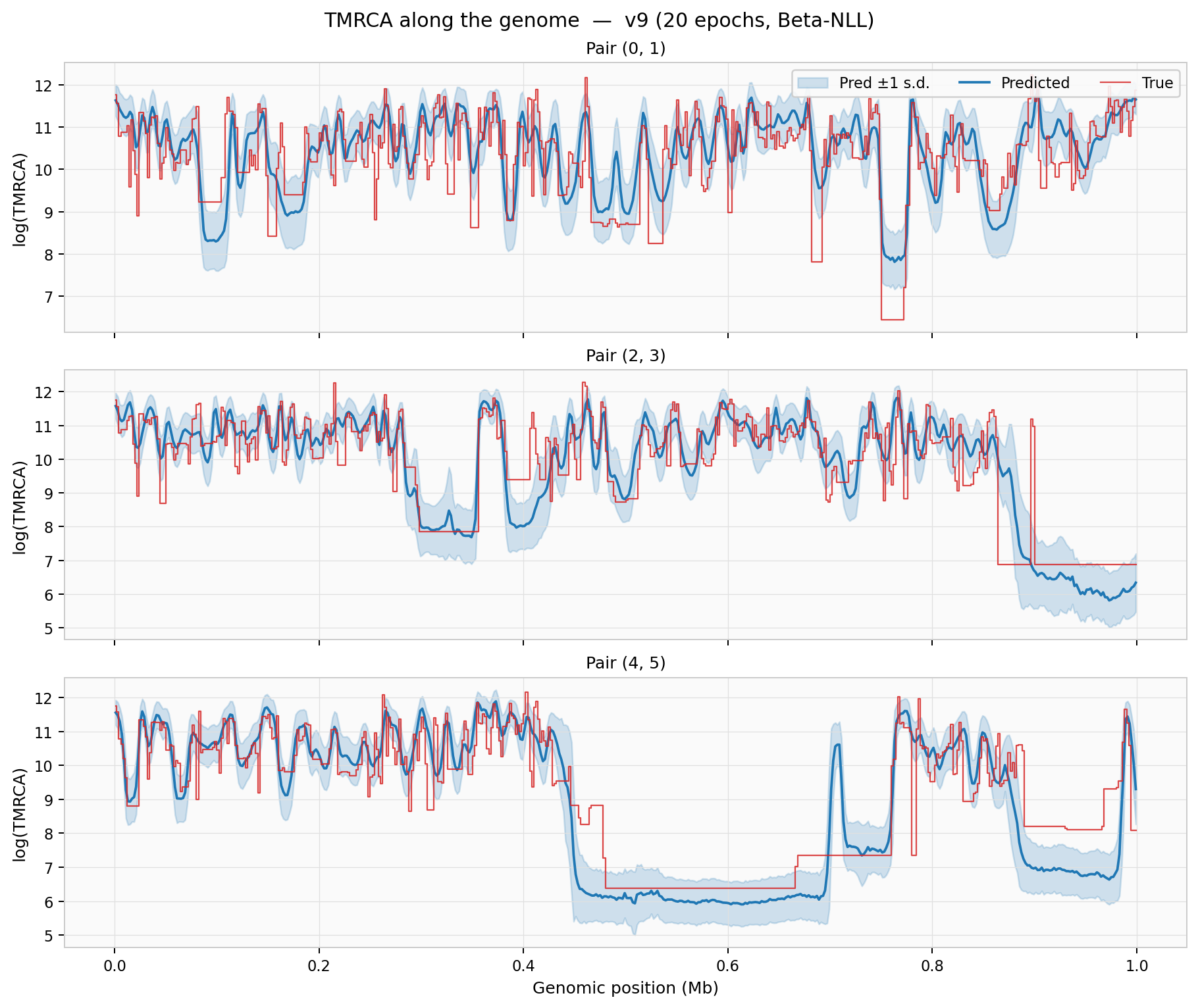
TMRCA along the genome — predicted mean (blue) with ±1 s.d. confidence band overlaid on the true values (red step) for three sample pairs:
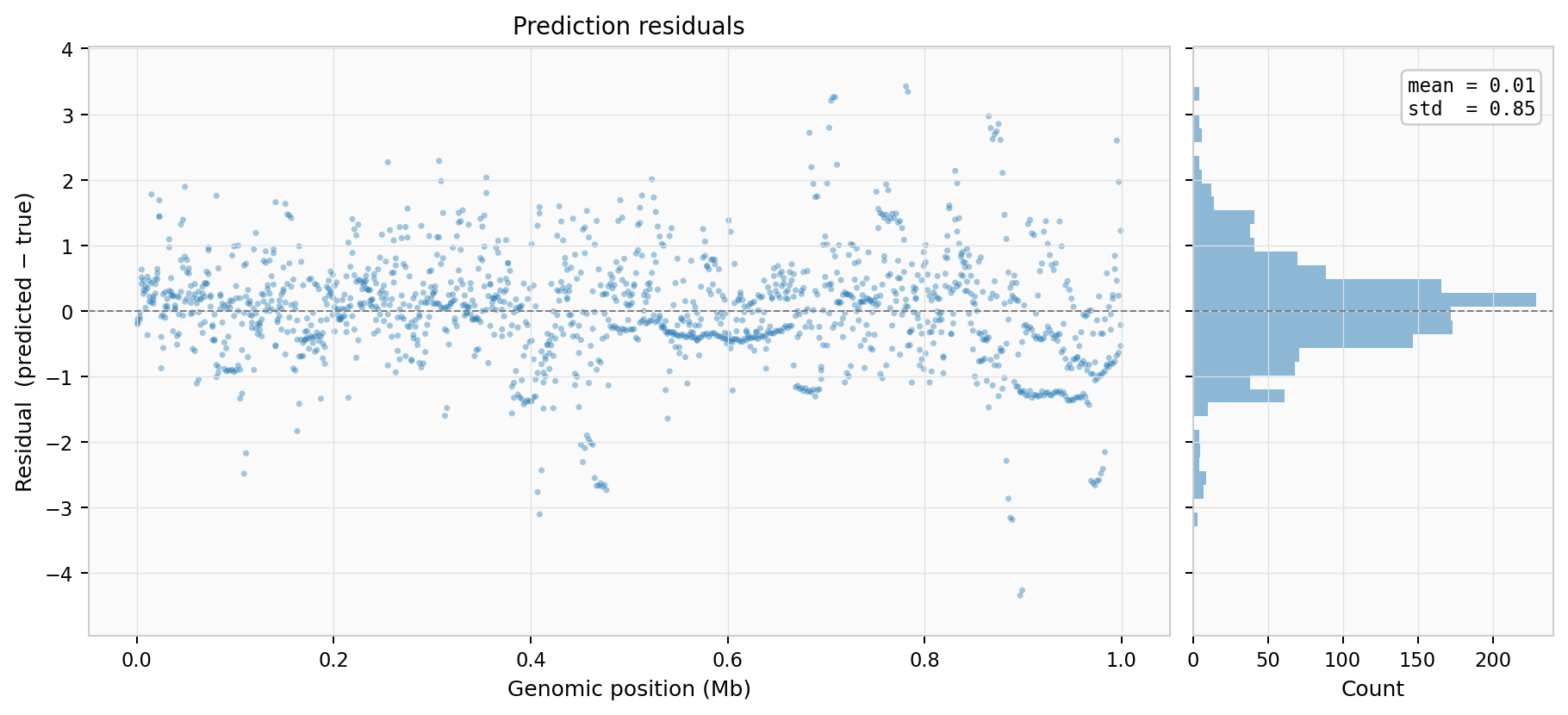
Residuals — spatial distribution and histogram of prediction errors:
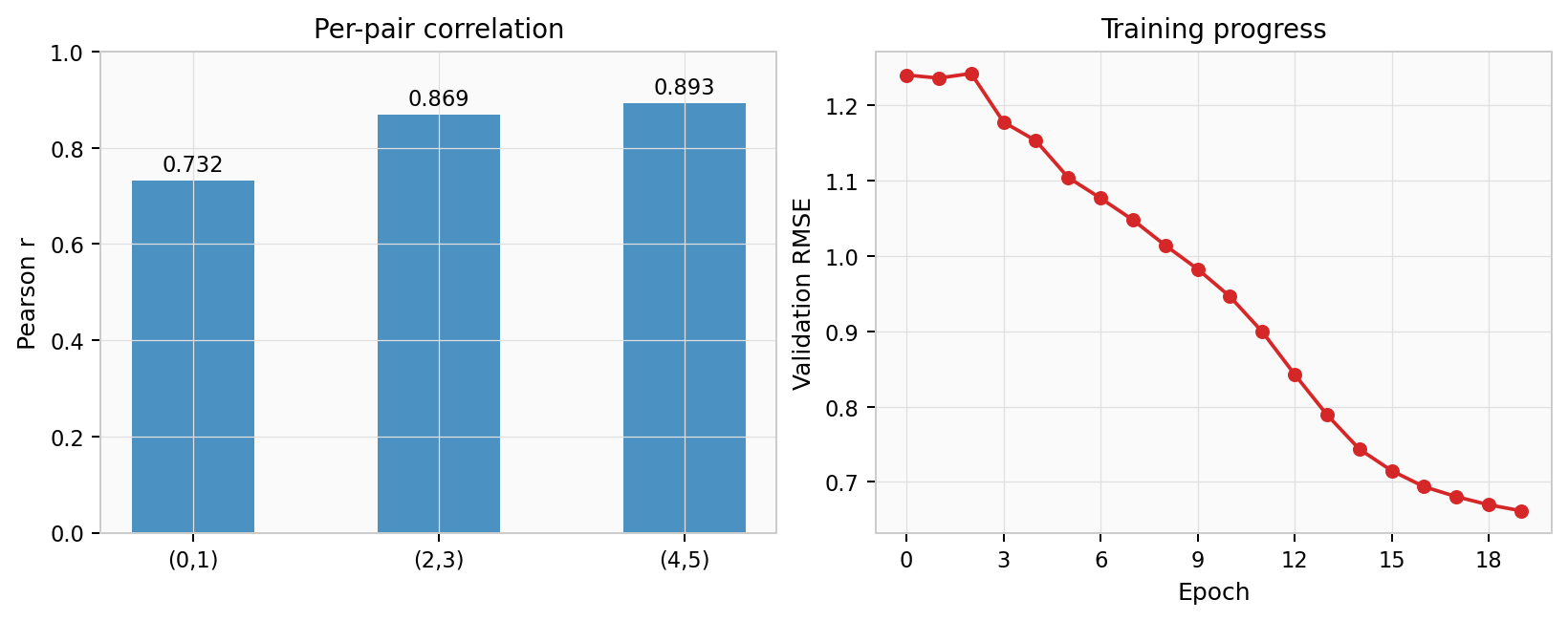
Summary — per-pair Pearson correlation and validation RMSE training curve: